




 V košíku
V košíku


V súčasnej dobe sa ukazuje, že jednou z mála funkčných liečebných metód ochorenia COVID-19, ktorá sa začína zavádzať do praxe, je využívanie krvnej plazmy vyliečených pacientov.

Leidenská mutace je vrozená dědičná porucha srážlivosti krve. Vzniká zvýšenou srážlivostí krve, pacient je tak náchylnější k tvorbě krevních sraženin, tzv. trombů. Rizikovou skupinou jsou mladé dívky s genetickou predispozicí, které užívají hormonální antikoncepci nebo kouří.
SKRYŤ POPIS
Mutace genu pro srážlivý faktor II (F II protrombin) je vrozená mutace. Je spojena se zvýšeným rizikem žilní trombózy.
SKRYŤ POPIS
Detekce chromozomových abnormalit probíhá kitem Panorama (Natera) a NGS sekvenováním.
SKRYŤ POPIS
Vrozená hluchota se vyskytuje zhruba u 1 z 1000 dětí, z toho přibližně 80 % případů je děděno autozomálně recesivní cestou. Nedávno bylo objeveno, že nejčastěji se vyskytuje u nosičů mutace genu GJB na 13. chromozomu. Tato mutace (s názvem 35delG) způsobuje v naší populaci přes 60 % případů vrozené hluchoty. Výskyt je často sledován v manželstvích neslyšících osob nebo u příbuzenských vztahů.
Test je doporučen pro všechny bez rozdílu, ještě před početím. U přenašečů s výše zmíněnou mutací je pak velmi doporučeno testování i jejich partnerů, protože riziko, že společné dítě bude také neslyšící, je poměrně vysoká (až 3 %).
Detekce mutace 35delG v genu pro Connexin 26 probíhá pomocí alelově specifické PCR (Poly Chain Reaction) metody.
SKRYŤ POPIS
Cystická fibróza je nejčastější dědičná metabolická vada v naší populaci, která se projevuje poruchou reprodukce u muže, ale i postupným chronickým onemocněním dýchacích cest a nedostatečnou sekrecí slinivky břišní. Jedná se o dědičné onemocnění způsobené mutací genu produkujícího protein CFTR (anglicky Cystic Fibrosis Transmembrane Conductance Regulator). Příčina cystické fibrózy je v současné době léčitelná, ale nedá se plně vyléčit a postižený trpí projevy celý život. V naší populaci toto onemocnění připadá na 1:2500–1:3000 narozených dětí.
Test zahrnuje 33 vyšetřovaných mutací, což znamená 91,7 % všech mutací CFTR genu v české populaci. Vyloučením těchto mutací se snižuje pravděpodobnost nosičství mutace v CFTR genu na 1:315 (0,32 %).
SKRYŤ POPIS
Spinální muskulární atrofie (SMA) je velmi časté dědičné (autozomálně recesivní) onemocnění, které způsobuje časem výraznou svalovou ochablost. Frekvence přenašečů mutace je 1:47 a SMA onemocní 1 z 10 000 dětí. Progres nemoci je individuální, ovlivňuje ho řada faktorů. Častá je ovšem dechová nedostatečnost, která se může stát později i příčinou smrti.
Vyšetření je proto doporučováno podstoupit ještě před početím, aby onemocnění nebylo přeneseno na potomka. Během testu na SMA se vyšetřuje gen, který kóduje SMN (Survival Motor Neuron) protein. Ten reguluje přežití motorických neuronů (inervujíc svaly) a jeho nedostatek způsobuje progresívní proximální svalovou slabost a atrofii.
Detekce počtu kopií SMN1 a SMN2 genu se provádí metodou MLPA (Multiplex Ligation-dependent Probe Amplification) pomocí kitu SALSA MLPA KIT P060-B2 SMA (MRC-Holland) na genetickém analyzátoru ABI3130 (Applied Biosystems).
SKRYŤ POPIS
V naší populaci onemocní rakovinou každý třetí a každý čtvrtý na ni zemře. V počtu onkologicky nemocných stále zaujímáme přední místa v Evropě a nádorové onemocnění má v rodině prakticky každý. Většinou se vyskytuje sporadicky, ačkoliv rizikové faktory, jako je vyšší věk či výskyt fyzikálních a chemických karcinogenů (látka způsobující rakovinné bujení) jsou zjevné. Nicméně 10 % nádorů, má dědičný charakter a je možné je velmi často vysledovat ještě před jejich propuknutím, a tak komplikacím předejít. Proto je nutné se včas řádně nechat otestovat a předejít tak závažným zdravotním komplikacím v pozdějších letech.
Pro účely testování na nejznámější onkologická onemocnění byl vyvinut tento balíček, který obsahuje test na známé genetické predispozice nádorů prsu či vaječníků, trávicího traktu a další mutace v genech, které běžně působí jako kontrola před rakovinovým bujením.
Asi nejznámější je dědičný nádor prsu či vaječníků. Nádory prsu u žen představují vysoké riziko, které od 30 let prokazatelně stoupá a nad 45 let představuje až 85 % všech nádorů. Příčinou je mutace v tumor-supresorových genech (genech regulujících buněčné dělení a tím potlačující růst nádoru) BRCA1 nebo BRCA2. Riziko vzniku tohoto nádoru je odhadováno zhruba na 85 % do věku 70 let a onemocnět jím mohou i muži. Riziko onemocnění nádorem vaječníků se u nosiček těchto mutací odhaduje až na 60 %.
Další mutacemi v genech, kterými lze vysledovat syndrom dědičného karcinomu prsu a vaječníků jsou: CHEK2, TP53, PALB2, BARD1, BRIP1 a ATM. Stejně jako proteiny genů BRCA1 a BRCA2, jsou tyto geny velmi důležité jako kontrola při buněčném dělení a následném růstu.
Velmi často se také setkáváme s nádory trávicího ústrojí (karcinom jícnu, nádory žaludku, tenkého střeva, tlustého střeva, konečníku, jater a rakovina slinivky). Pro tyto případy byly vybrány k testování mutace v genech: MHL1, MHS2, jejichž mutace jsou často spojeny s rakovinou tlustého střeva, konkrétně s hereditárním nepolypózním karcinomem tlustého střeva (HNPCC, Lynchův syndrom). A dále geny MSH6, EPCAM, MUTYH, APC a z nich vznikající proteiny, které hrají esenciální roli v opravě DNA, při růstu a dělení buněk, regulují aktivitu ostatních genů a důležitých procesů správného dělení buněk a tkání a zamezují tak vzniku nádoru.
Zbylé testované geny, jako je CDH1, PMS2, PTEN, RAD50, RAD51C, RAD51D, STK11 a NBN u nichž se mutace testují, jsou obecně spojené s častými výskyty rakoviny žaludku, prsu, štítné žlázy či vaječníků, kolorektálním karcinomem a dalšími, často se vyskytujícími nádory.
Mutační analýza genů probíhá metodou masivně paralelního sekvenování (platforma MiSeq, Illumina).
SKRYŤ POPIS
Mikrodelece chromozomu Y u mužů je příčinou až 10 % těžkých forem neplodnosti. Delece (ztráta) genů z oblasti a, b, c AZF (Azoospermia Factor) na dlouhém raménku mají za následek špatný vývoj spermií. Nejzávažnější jsou delece v oblasti AZFa, které mají často za následek úplné chybění spermatogonií (mužské zárodečné buňky). Oproti tomu delece oblasti AZFb a AZFc má většinou azoospermický až oligozoospermický projev. Klasické cytogenetické metody však tyto malé chromozomální změny neodhalí, a proto je při neplodnosti velmi doporučováno toto vyšetření podstoupit, aby se případné nedostatky odhalily a pomocí metod asistované reprodukce jim mohlo být umožněno mít děti (po chirurgické extrakci spermií z varlete nebo nadvarlete - TESE, MESA, PESA).
SKRYŤ POPIS
Vyšetření Gilbertův syndrom - probíhá pomocí PCR amplifikace části promotorové oblasti UGT 1A1 genu zahrnující TATA box a následně fragmentační analýzy na genetickém analyzátoru ABI PRISM 3130.
SKRYŤ POPIS
Cytogenetické vyšetření karyotypu (chromozomová výbava) je důležitou součástí prekoncepčního vyšetření (=vyšetření před početím) neplodných. Test je zaměřen na chromosomové vybavení buňky, za jehož pomoci můžeme vyloučit nosičství strukturních aberací (zlomů v chromozomech), které pak většinou znamenají riziko závažných změn genetické výbavy postihující řadu orgánů u plodu. V současnosti neexistuje ani účinná prevence, ani cílená léčba chromozomálních vad a životní prognóza postižených je často nepříznivá.
Doporučené jsou zejména pro ženy a muže:
Normální karyotyp ženy je 46, XX, muže 46, XY.
SKRYŤ POPIS
Metoda fluorescenční in situ hybridizace (FISH) se používá k upřesnění a doplnění chromosomálního vyšetření. FISH odhalí i malé mozaiky početní aberace. Vyšetření probíhá s centromerickou sondou pro pohlavní chromosom X a výsledkem je vyhodnocení minimálně 100 jader v interfázi (= období mezi dvěma děleními buňky) na jeden mikroskopický preparát ve fluorescenčním mikroskopu.
SKRYŤ POPIS
Array CGH (komparativní genomová hybridizace na čipu) je molekulárně cytogenetické vyšetření celého genomu. Slouží k odhalení nebalancovaných aberací (ztrát a zisků sekvencí), jejichž umístění v genomu není předem známé. Metoda může odhalit příčinu azoospermie nebo těžké oligoastenozoospermie.
SKRYŤ POPIS
Syndrom fragilního chromozómu X (Martin-Bell syndrom) je jednou z nejčastějších příčin genetického mentálního postižení, a to zejména u mužů. Ženy trpí onemocněním mnohem méně, protože onemocnění je vázané na chromozom X. U žen se tak může ale i nemusí onemocnění vůbec projevit, protože mají chromozomy XX, riziko propuknutí je pod 50 %. To ale neplatí u mužů, kteří nesou chromozomy XY, zde je jisté, že onemocnění propukne.
Onemocnění působí svému nositeli značné komplikace. Tato mutace vede k různým stupňům duševního deficitu, který se může pohybovat od lehčích poruch (např. učení) až po těžkou mentální retardaci. Onemocnění je hned po Downově syndromu nejčastější příčinou přenášeného mentálního zaostávání (IQ 20-70).
U nosičů mutace často pozorujeme také typický vzhled s dysmorfickými rysy (protáhlý obličej s prominující bradou a velkýma odstávajícíma ušima), u mužů se vyskytuje makroorchidismus (abnormální zvětšení jednoho nebo obou varlat) a dále rozdílné chování. Bývají popisovány různě závažné poruchy chování, jako jsou poruchy učení, autismus či hyperaktivita. Syndrom se vyskytuje přibližně s frekvencí 1:3000-1:4000 u mužů a 1:8000 u žen.
SKRYŤ POPIS
Při testu otcovství se porovnává genetická příbuznost dítěte a předpokládaného otce. Vyloučení otcovství je stanoveno s jistotou, potvrzení otcovství je s pravděpodobností nad 99,5 %. Výsledek je informace pro objednavatele, neslouží jako listinný důkaz u soudu.
Určení otcovství probíhá pomocí hypervariabilních STR markerů (neboli Short Tandem Repeat sekvencí) lidské DNA.
Laktózová intolerance - Vyšetření laktózové intolerance probíhá pomocí stanovení genotypu v pozici 13910 v regulační oblasti LCT genu (genu kódujícím enzym laktázu) metodou SSP-PCR (Single Specific Primer-Polymerase Chain Reaction).
Nosiči genotypu T/T se projevují jako laktózově tolerantní i v dospělém věku. Nosiči genotypu C/C jsou laktózově tolerantní pouze v dětství, v dospělosti jsou laktózově intolerantní. Nosiči genotypu C/T jsou laktózově tolerantní, ale během onemocnění nebo stresu mohou projevovat nižší toleranci laktózy.
SKRYŤ POPIS
Test na celiakii zahrnuje vyšetření predispozičních HLA alel DQ2 a DQ8.
SKRYŤ POPIS
Majte svoje zdravie pod kontrolou priamo vo svojom smartfóne.
Aplikácia NEXTLIFE je teraz dostupná ZDARMA v App Store aj v Google Play.



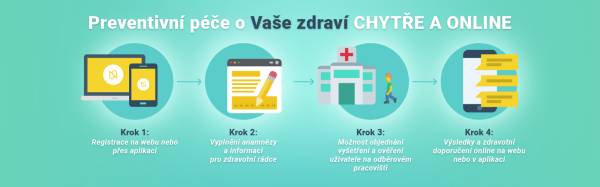
 Mobilná aplikácia
Mobilná aplikácia Ako to funguje
Ako to funguje Odberné miesta
Odberné miesta